Built-in analysis tools¶
As of version 0.14, the tools have a standardized output that is different from previous versions. Requested –columns will come first followed by a standard set of columns:
- variant_id - unique id from the databse
- family_id - family id for this row
- family_members - which family members were tested
- family_genotypes - genotypes of this family
- samples - samples contributing to this row appearing in the results
- family_count - number of families with this effect
Other tools such as mendel_errors additional columns at the end.
When queries are limited to variants in genes, the output will be in gene, chrom order as opposed to the usual gene, position order.
Note
As of version 0.16.0, the inheritance tools (autosomal_dominant, autosomal_recessive, comp_het, mendel_errors, de_novo) are now more strict by default.
A –lenient flag allows, e.g. allows some samples to be of unknown phenotype or to not have both parents of known phenotype.
The –allow-unaffected flag will result in reporting variants where unaffected samples share the same variants. The default will only report variants that are unique to affected samples.
The design decisions for this change are described here: https://github.com/arq5x/gemini/issues/388 a visual representation is here: https://github.com/arq5x/gemini/blob/master/inheritance.ipynb
Note
Candidate variants reported by the built-in inheritance model tools will appear in order by chromosome, then alphabetically by gene. In other words, they will not be in strict positional order for each chromosome. This is in an effort to group all candidate variants by gene since the gene is typically the atomic unit of interest.
Additionally, all current built-in tools (autosomal_dominant, comp_hets, and autosomal_recessive) only analyze autosomal (non sex chromosome) variants. Analysis of X- and Y-linked phenotypes must be done manually with –gt-filter
common_args: common arguments¶
The inheritance tools share a common set of arguments. We will describe them here and refer to them in the corresponding sections:
--columns¶
This flag is followed by a comma-delimited list of columns the user is requestin in the output.
--min-kindreds 1¶
This is the number of families required to have a variant in the same gene in order for it to be reported. For example, we may only be interested in candidates where at least 4 families have a variant in that gene.
--families¶
By default, candidate variants are reported for all families in the database.
One can restrict the analysis to variants in specific familes with the
--families option. Families should be provided as a comma-separated list
--filter¶
By default, each tool will report all variants regardless of their putative
functional impact. In order to apply additional constraints on the variants
returned, one can use the --filter option. Using SQL syntax, conditions
applied with the --filter options become WHERE clauses in the query issued to
the GEMINI database.
-d [0] (depth)¶
Filter variants that do not have at least this depth for all members in a a family. Default is 0.
--min-gq [0]¶
Filter variants that do not have at least this genotype quality for each sample in a family. Default is 0. Higher values are more stringent.
--allow-unaffected¶
By default, candidates that also appear in unaffected samples are not reported if this flag is specified, such variants will be reported.
--lenient¶
Loosen the restrictions on family structure. This will allow, for example, finding compound_hets in unaffected samples.
--gt-pl-max¶
In order to eliminate less confident genotypes, it is possible to enforce a maximum PL value for each sample. On this scale, lower values indicate more confidence that the called genotype is correct. 10 is a reasonable value. This is applied per-family such that all members of a family must meet this level in order to by reported in the final results.
comp_hets: Identifying potential compound heterozygotes¶
Many autosomal recessive disorders are caused by compound heterozygotes. Unlike canonical recessive sites where the same recessive allele is inherited from both parents at the _same_ site in the gene, compound heterozygotes occur when the individual’s phenotype is caused by two heterozygous recessive alleles at _different_ sites in a particular gene.
We are looking for two (typically loss-of-function (LoF)) heterozygous variants impacting the same gene at different loci. The complicating factor is that this is _recessive_ and as such, we must also require that the consequential alleles at each heterozygous site were inherited on different chromosomes (one from each parent). Where possible, comp_hets will phase by transmission. Once this has been done, the comp_hets tool will provide a report of candidate compound heterozygotes for each sample/gene.
Non-exonic/non-coding analyses: comp_hets excludes intronic/non-coding variants for which impact_severity == ‘LOW’ AND is_exonic == FALSE. Therefore, comp_hets will not retrieve most pairs of variants that are downstream or upstream of a gene or are intronic unless otherwise annotated with medium or high impact_severity.
Note
As of version 0.16.0 the comp_het tool will perform family-based phasing
by default in order to provide better candidates even in the absence of
unphased genotypes. Any candidate that could be one element of a comp_het
will also be phaseable as long as the parents and their genotypes are known.
As of version 0.16.1, the –ignore-phasing option is removed and there is no –lenient option.
In 0.16.2, a –pattern-only flag was added to find compound hets by inheritance pattern without regard to affection status. A priority code was also added where variants with priority 1 are much more informative. See docs below for further information.
Genotype Requirements¶
- All affected individuals must be heterozygous at both sites.
- No unaffected can be homozygous alterate at either site.
- Neither parent of an affected sample can be homozygous reference at both sites.
- If any unphased-unaffected is het at both sites, the site will be give lower priority
- No phased-unaffected can be heterozygous at both sites.
- –allow-unaffected keeps sites where a phased unaffected shares the het-pair
- unphased, unaffected that share the het pair are counted and reported for each candidate pair.
- Remove candidates where an affected from the same family does NOT share the same het pair.
- Sites are automatically phased by transmission when parents are present in order to remove false positive candidates.
- If data from one or both parents are unavailable and the child’s data was not phased prior to loading into GEMINI, all comp_het variant pairs will automatically be given at most priority == 2. If there’s only a single parent and both the parent and the affected are HET at both sites, the candidate will have priority 3.
- –max-priority x can be used to set the maximum allowed priority level at which candidate pairs are included in the output.
we prioritize with these rules:
| mom | dad | kid | phaseable | priority | notes |
|---|---|---|---|---|---|
| R-H | H-R | H-H | both | 1 | both sites phaseable and alts on opposite chroms |
| n/a | n/a | H-H | NO | 2 | singleton (unphaseable) HETs have priority 2. |
| R-H | H-H | H-H | one | 3 | should be a rare occurrence |
| H-H | H-H | H-H | NO | 3 | should be a rare occurrence |
| H-H | UNK | H-H | NO | 3 | missing parent and all hets. |
| A-R | H-H | H-H | both | NA | exclude hom-alts from un-affecteds |
| R-R | H-H | H-H | both | NA | phaseable, but alts are on the same chroms. |
Note
candidates of priority == 3 are very unlikely (< 1%) to be causal for a rare Mendelian condition (see: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3734130/); we report them for completeness, but strongly recommend using priority 1 and 2 only. Priority 2 is useful when there are multiple families, some of which consist of only a single sequenced, affected sample.
Pattern Only¶
To find compound heterozygotes by inheritance pattern only, without regard to affections, the following rules are used (with –pattern-only):
- Kid must be HET at both sites.
- Kid must have alts on different chromosomes.
- Neither parent can be HOM_ALT at either site.
- If either parent is phased at both sites and matches the kid, it’s excluded.
- If either parent is HET at both sites, priority is reduced.
- When the above criteria are met, and both parents and kid are phased or parents are HET at different sites, the priority is 1.
- If both parents are not phased, the priority is 2.
- For every parent that’s a het at both sites, the priority is incremented by 1.
- The priority in a family is the minimum found among all kids.
Note
Each pair of consecutive lines in the output represent the two variants for a compound heterozygote in a give sample. The third column, comp_het_id, tracks the distinct compound heterozygote variant pairs.
Example usage with a subset of columns:
$ gemini comp_hets my.db --columns "chrom, start, end" test.comp_het_default.2.db
chrom start end gene alt variant_id family_id family_members family_genotypes samples family_count comp_het_id
chr1 17362 17366 WASH7P T 1 3 dad_3(dad;unaffected),mom_3(mom;unaffected),child_3(child;affected) TTCT|T,TTCT|TTCT,TTCT|T child_3 2 1
chr1 17729 17730 WASH7P A 2 3 dad_3(dad;unaffected),mom_3(mom;unaffected),child_3(child;affected) C|A,C|A,A|C child_3 2 1
chr1 17362 17366 WASH7P T 1 4 dad_4(dad;unaffected),mom_4(mom;unaffected),child_4(child;affected) TTCT|T,TTCT|TTCT,TTCT|T child_4 2 1
chr1 17729 17730 WASH7P A 2 4 dad_4(dad;unaffected),mom_4(mom;unaffected),child_4(child;affected) C|A,C|A,A|C child_4 2 1
This indicates that samples child_3 and child_4 have a candidate compound heterozygotes in WASH.
the following command would further restrict candidate genes to those genes with a compound heterozygote in at least two families:
$ gemini comp_hets -d 50 \
--columns "chrom, start, end, ref, alt" \
--filter "impact_severity = 'HIGH'" \
--allow-unaffected \
--min-kindreds 2 \
my.db
Now, this does not require that the family members are necessarily restricted to solely
those that are affected. To impose this restriction, we remove the --allow-unaffected
flag
$ gemini comp_hets -d 50 \
--columns "chrom, start, end, ref, alt" \
--filter "impact_severity = 'HIGH'" \
--min-kindreds 2 \
my.db
We may also specify the families of interest:
$ gemini comp_hets --families 1 my.db
$ gemini comp_hets --families 1,7 my.db
gene-where¶
The default selection of genes is by the clause: “is_exonic = 1 or impact_severity != ‘LOW’” This can be specified to limit to a different subset, e.g. “gene != ‘’
mendelian_error: Identify non-mendelian transmission.¶
Note
This tool requires that you identify familial relationships via a PED file when loading your VCF into gemini via:
gemini load -v my.vcf -p my.ped my.db
We can query for mendelian errors in trios including:
- loss of heterozygosity
- implausible de-novo mutations
- de-novo mutations
- uniparental disomy
Genotype Requirements¶
- (LOH) kind and one parent are opposite homozygotes; other parent is HET
- (uniparental disomy) parents are opposite homozygotes; kid is homozygote;
- (plausible de novo) kid is het. parents are same homozygotes
- (implausible de novo) kid is homozygoes. parents are same homozygotes and opposite to kid.
If allow –only-affected is used, then the tools will only consider samples that have parents and are affected. The default is to consider any sample with parents.
This tool will report the probability of a mendelian error in the final column that is derived from the genotype likelihoods if they are available.
Example:
$ gemini mendel_errors --columns "chrom,start,end" test.mendel.db --gt-pl-max 1
chrom start end variant_id family_id family_members family_genotypes samples family_count violation violation_prob
chr1 10670 10671 1 CEPH1463 NA12889(dad;unknown),NA12890(mom;unknown),NA12877(child;unknown) G/G,G/G,G/C NA12877 1 plausible de novo 0.962
chr1 28493 28494 2 CEPH1463 NA12889(dad;unknown),NA12890(mom;unknown),NA12877(child;unknown) T/C,T/T,C/C NA12877 1 loss of heterozygosity 0.660
chr1 28627 28628 3 CEPH1463 NA12889(dad;unknown),NA12890(mom;unknown),NA12877(child;unknown) C/C,C/C,C/T NA12877 1 plausible de novo 0.989
chr1 267558 267560 5 CEPH1463 NA12889(dad;unknown),NA12890(mom;unknown),NA12877(child;unknown) C/C,C/C,CT/C NA12877 1 plausible de novo 0.896
chr1 537969 537970 7 CEPH1463 NA12889(dad;unknown),NA12890(mom;unknown),NA12877(child;unknown) C/C,C/C,C/T NA12877 1 plausible de novo 0.928
chr1 547518 547519 11 CEPH1463 NA12889(dad;unknown),NA12890(mom;unknown),NA12877(child;unknown) G/G,G/G,G/T NA12877 1 plausible de novo 1.000
chr1 589081 589086 14 CEPH1463 NA12889(dad;unknown),NA12890(mom;unknown),NA12877(child;unknown) G/G,GAGAA/GAGAA,G/G NA12877 1 uniparental disomy 0.940
chr1 749688 749689 16 CEPH1463 NA12889(dad;unknown),NA12890(mom;unknown),NA12877(child;unknown) T/T,T/T,G/G NA12877 1 implausible de novo 0.959
chr1 788944 788945 17 CEPH1463 NA12889(dad;unknown),NA12890(mom;unknown),NA12877(child;unknown) C/C,G/G,G/G NA12877 1 uniparental disomy 0.914
chr1 1004248 1004249 22 CEPH1463 NA12889(dad;unknown),NA12890(mom;unknown),NA12877(child;unknown) G/G,G/G,G/C NA12877 1 plausible de novo 1.000
Where, here, we have required the called genotype to have at most a PL of 1 (lower is more confident). Note that the “violation” column indicates the type of mendelian error and the final column can be used for further filtering, with higher numbers indicating a greater probability of mendelian error. We have found > 0.99 to be a reasonable cutoff.
Arguments are similar to the other tools:
positional arguments:
db The name of the database to be queried.
optional arguments:
-h, --help show this help message and exit
--columns STRING A list of columns that you would like returned. Def. =
"*"
--filter STRING Restrictions to apply to variants (SQL syntax)
--min-kindreds MIN_KINDREDS
The min. number of kindreds that must have a candidate
variant in a gene.
--families FAMILIES Restrict analysis to a specific set of 1 or more
(comma) separated) families
-d MIN_SAMPLE_DEPTH The minimum aligned sequence depth required for
each sample in a family (default = 0)
--gt-pl-max GT_PHRED_LL
The maximum phred-scaled genotype likelihod (PL)
allowed for each sample in a family.
--allow-unaffected consider candidates that also appear in unaffected samples.
de_novo: Identifying potential de novo mutations.¶
Note
1. This tool requires that you identify familial relationships via a PED file when loading your VCF into gemini via:
gemini load -v my.vcf -p my.ped my.db
Genotype Requirements¶
- all affecteds must be het
- [affected] all unaffected must be homref or homalt
- at least 1 affected kid must have unaffected parents
- [strict] if an affected has affected parents, it’s not de_novo
- [strict] all affected kids must have unaffected (or no) parents
- [strict] warning if none of the affected samples have parents.
The last 3 items, prefixed with [strict] can be turned off with –lenient
If –allow-unaffected is specified, then the item prefixed [affected] is not required.
Example PED file format for GEMINI
#Family_ID Individual_ID Paternal_ID Maternal_ID Sex Phenotype Ethnicity
1 S173 S238 S239 1 2 caucasian
1 S238 -9 -9 1 1 caucasian
1 S239 -9 -9 2 1 caucasian
2 S193 S230 S231 1 2 caucasian
2 S230 -9 -9 1 1 caucasian
2 S231 -9 -9 2 1 caucasian
3 S242 S243 S244 1 2 caucasian
3 S243 -9 -9 1 1 caucasian
3 S244 -9 -9 2 1 caucasian
4 S253 S254 S255 1 2 caucasianNEuropean
4 S254 -9 -9 1 1 caucasianNEuropean
4 S255 -9 -9 2 1 caucasianNEuropean
Assuming you have defined the familial relationships between samples when loading your VCF into GEMINI, one can leverage a built-in tool for identifying de novo (a.k.a spontaneous) mutations that arise in offspring.
example¶
$ gemini de_novo --columns "chrom,start,end" test.de_novo.db
chrom start end variant_id family_id family_members family_genotypes samples family_count
chr10 1142207 1142208 1 1 1_dad(dad;unaffected),1_mom(mom;unaffected),1_kid(child;affected) T/T,T/T,T/C 1_kid 1
chr10 48003991 48003992 2 2 2_dad(dad;unaffected),2_mom(mom;unaffected),2_kid(child;affected) C/C,C/C,C/T 2_kid 1
chr10 48004991 48004992 3 3 3_dad(dad;unaffected),3_mom(mom;unaffected),3_kid(child;affected) C/C,C/C,C/T 3_kid 1
chr10 135336655 135336656 4 4 1_dad(dad;unaffected),1_mom(mom;unaffected),1_kid(child;affected) G/G,G/G,G/A 1_kid 2
chr10 135336655 135336656 4 4 2_dad(dad;unaffected),2_mom(mom;unaffected),2_kid(child;affected) G/G,G/G,G/A 2_kid 2
chr10 135369531 135369532 5 5 1_dad(dad;unaffected),1_mom(mom;unaffected),1_kid(child;affected) T/T,T/T,T/C 1_kid 3
chr10 135369531 135369532 5 5 2_dad(dad;unaffected),2_mom(mom;unaffected),2_kid(child;affected) T/T,T/T,T/C 2_kid 3
chr10 135369531 135369532 5 5 3_dad(dad;unaffected),3_mom(mom;unaffected),3_kid(child;affected) T/T,T/T,T/C 3_kid 3
Note
The output will always start with the the requested columns followed by the 5 columns enumerated at the start of this document.
$ gemini de_novo -d 50 --columns "chrom,start,end" test.de_novo.db
chrom start end variant_id family_id family_members family_genotypes samples family_count
chr10 135369531 135369532 5 5 3_dad(dad;unaffected),3_mom(mom;unaffected),3_kid(child;affected) T/T,T/T,T/C 3_kid 1
example¶
if we wanted to restrict candidate variants to solely those with a HIGH predicted functional consequence, we could use the following:
$ gemini de_novo \
--columns "chrom, start, end, ref, alt" \
--filter "impact_severity = 'HIGH'" \
test.de_novo.db
chrom start end ref alt variant_id family_id family_members family_genotypes samples family_count
chr10 1142207 1142208 T C 1 1 1_dad(dad;unaffected),1_mom(mom;unaffected),1_kid(child;affected) T/T,T/T,T/C 1_kid 1
example¶
the following command would further restrict candidate genes to those genes with a de novo variant in at least two families:
$ gemini de_novo \
--columns "chrom, start, end, ref, alt" \
--filter "impact_severity = 'HIGH'" \
--min-kindreds 2 \
test.de_novo.db
example¶
By default, candidate de novo variants are reported for families
in the database. One can restrict the analysis to variants in
specific familes with the --families option. Families should be provided
as a comma-separated list
$ gemini de_novo --families 1 my.db
$ gemini de_novo --families 1,7 my.db
autosomal_recessive: Find variants meeting an autosomal recessive model.¶
Warning
By default, this tool requires that you identify familial relationships via a PED file when loading your VCF into GEMINI. For example:
gemini load -v my.vcf -p my.ped my.db
However, in the absence of established parent/child relationships in the PED file, GEMINI will issue a WARNING, yet will attempt to identify autosomal recessive candidates for all samples marked as “affected”.
Genotype Requirements¶
- all affecteds must be hom_alt
- [affected] no unaffected can be hom_alt (can be unknown)
- [strict] if parents exist they must be unaffected and het for all affected kids
- [strict] if there are no affecteds that have a parent, a warning is issued.
if –lenient is specified, the 2 points prefixed with “[strict]” are not required.
if –allow-unaffected is specified, the point prefix with “[affected]” is not required.
default behavior¶
Assuming you have defined the familial relationships between samples when loading your VCF into GEMINI, one can leverage a built-in tool for identifying variants that meet an autosomal recessive inheritance pattern. The reported variants will be restricted to those variants having the potential to impact the function of affecting protein coding transcripts.
For the following examples, let’s assume we have a PED file for 3 different families as follows (the kids are affected in each family, but the parents are not):
$ cat families.ped
1 1_dad 0 0 -1 1
1 1_mom 0 0 -1 1
1 1_kid 1_dad 1_mom -1 2
2 2_dad 0 0 -1 1
2 2_mom 0 0 -1 1
2 2_kid 2_dad 2_mom -1 2
3 3_dad 0 0 -1 1
3 3_mom 0 0 -1 1
3 3_kid 3_dad 3_mom -1 2
$ gemini autosomal_recessive test.auto_rec.db --columns "chrom,start,end,gene"
chrom start end gene variant_id family_id family_members family_genotypes samples family_count
chr10 48003991 48003992 ASAH2C 2 2 1_dad(dad;unaffected),1_mom(mom;unaffected),1_kid(child;affected) C/T,C/T,T/T 1_kid 1
chr10 48004991 48004992 ASAH2C 3 3 2_dad(dad;unaffected),2_mom(mom;unaffected),2_kid(child;affected) C/T,C/T,T/T 2_kid 1
chr10 135369531 135369532 SYCE1 5 5 3_dad(dad;unaffected),3_mom(mom;unaffected),3_kid(child;affected) T/C,T/C,C/C 3_kid 1
chr10 1142207 1142208 WDR37 1 1 1_dad(dad;unaffected),1_mom(mom;unaffected),1_kid(child;affected) T/C,T/C,C/C 1_kid 2
chr10 1142207 1142208 WDR37 1 1 2_dad(dad;unaffected),2_mom(mom;unaffected),2_kid(child;affected) T/C,T/C,C/C 2_kid 2
Note
The output will always start with the requested columns and end with the 5 extra columns enumerated at the start of this document.
To restrict the report to genes with variants (doesn’t have to be the _same_ variant) observed in at least two kindreds, use the following:
$ gemini autosomal_recessive \
--columns "gene, chrom, start, end, ref, alt, impact, impact_severity" \
--min-kindreds 2 \
test.auto_rec.db
gene chrom start end ref alt impact impact_severity variant_id family_id family_members family_genotypes samples family_count
ASAH2C chr10 48003991 48003992 C T non_syn_coding MED 2 2 1_dad(dad;unaffected),1_mom(mom;unaffected),1_kid(child;affected) C/T,C/T,T/T 1_kid 1
ASAH2C chr10 48004991 48004992 C T non_syn_coding MED 3 3 2_dad(dad;unaffected),2_mom(mom;unaffected),2_kid(child;affected) C/T,C/T,T/T 2_kid 1
WDR37 chr10 1142207 1142208 T C stop_loss HIGH 1 1 1_dad(dad;unaffected),1_mom(mom;unaffected),1_kid(child;affected) T/C,T/C,C/C 1_kid 2
WDR37 chr10 1142207 1142208 T C stop_loss HIGH 1 1 2_dad(dad;unaffected),2_mom(mom;unaffected),2_kid(child;affected) T/C,T/C,C/C 2_kid 2
to report only those with a HIGH predicted functional consequence, we could use the following:
$ gemini autosomal_recessive \
--columns "gene, chrom, start, end, ref, alt, impact, impact_severity" \
--min-kindreds 2 \
--filter "impact_severity = 'HIGH'" \
test.auto_rec.db
gene chrom start end ref alt impact impact_severity variant_id family_id family_members family_genotypes samples family_count
WDR37 chr10 1142207 1142208 T C stop_loss HIGH 1 1 1_dad(dad;unaffected),1_mom(mom;unaffected),1_kid(child;affected) T/C,T/C,C/C 1_kid 2
WDR37 chr10 1142207 1142208 T C stop_loss HIGH 1 1 2_dad(dad;unaffected),2_mom(mom;unaffected),2_kid(child;affected) T/C,T/C,C/C 2_kid 2
To limit to confidently called genotypes:
$ gemini autosomal_dominant \
--columns "gene, chrom, start, end, ref, alt, impact, impact_severity" \
--filter "impact_severity = 'HIGH'" \
--min-kindreds 1 \
--gt-pl-max 10 \
my.db
autosomal_dominant: Find variants meeting an autosomal dominant model.¶
Warning
0. version 0.16.0 changes the behavior of this tool to be more strict. To regain more lenient behavior, specify –lenient and –allow-unaffected.
By default, this tool requires that you identify familial relationships via a PED file when loading your VCF into GEMINI. For example:
gemini load -v my.vcf -p my.ped my.db
Genotype Requirements¶
- All affecteds must be het
- [affected] No unaffected can be het or homalt (can be unknown)
- de_novo mutations are not auto_dom (at least not in the first generation)
- At least 1 affected must have 1 affected parent (or have no parents).
- If no affected has a parent, a warning is issued.
- [strict] All affecteds must have parents with known phenotype.
- [strict] All affected kids must have at least 1 affected parent
If –lenient is specified, the items prefixed with “[strict]” are not required.
If –allow-unaffected is specified, the item prefix with “[affected]” is not required.
Note that for autosomal dominant –lenient allows singleton affecteds to be used to meet the –min-kindreds requirement if they are HET.
If there is incomplete penetrance in the kindred (unaffected obligate carriers), these individuals currently must be coded as having unknown phenotype or as being affected.
default behavior¶
For the following examples, let’s assume we have a PED file for 3 different families as follows (the kids are affected in each family, but the parents are not):
$ cat families.ped
1 1_dad 0 0 -1 1
1 1_mom 0 0 -1 1
1 1_kid 1_dad 1_mom -1 2
2 2_dad 0 0 -1 1
2 2_mom 0 0 -1 2
2 2_kid 2_dad 2_mom -1 2
3 3_dad 0 0 -1 2
3 3_mom 0 0 -1 -9
3 3_kid 3_dad 3_mom -1 2
$ gemini autosomal_dominant test.auto_dom.db --columns "chrom,start,end,gene"
chrom start end gene variant_id family_id family_members family_genotypes samples family_count
chr10 48003991 48003992 ASAH2C 3 3 2_dad(dad;unaffected),2_mom(mom;affected),2_kid(child;affected) C/C,C/T,C/T 2_mom,2_kid 2
chr10 48004991 48004992 ASAH2C 4 4 2_dad(dad;unaffected),2_mom(mom;affected),2_kid(child;affected) C/C,C/T,C/T 2_mom,2_kid 2
chr10 48003991 48003992 ASAH2C 3 3 3_dad(dad;affected),3_mom(mom;unknown),3_kid(child;affected) C/T,C/C,C/T 3_dad,3_kid 2
chr10 48004991 48004992 ASAH2C 4 4 3_dad(dad;affected),3_mom(mom;unknown),3_kid(child;affected) C/T,C/C,C/T 3_dad,3_kid 2
chr10 135336655 135336656 SPRN 5 5 3_dad(dad;affected),3_mom(mom;unknown),3_kid(child;affected) G/A,G/G,G/A 3_dad,3_kid 1
chr10 1142207 1142208 WDR37 1 1 2_dad(dad;unaffected),2_mom(mom;affected),2_kid(child;affected) T/T,T/C,T/C 2_mom,2_kid 2
chr10 1142207 1142208 WDR37 1 1 3_dad(dad;affected),3_mom(mom;unknown),3_kid(child;affected) T/C,T/T,T/C 3_dad,3_kid 2
$ gemini autosomal_dominant \
--columns "gene, chrom, start, end, ref, alt, impact, impact_severity" \
--min-kindreds 2 \
test.auto_dom.db
gene chrom start end ref alt impact impact_severity variant_id family_id family_members family_genotypes samples family_count
ASAH2C chr10 48003991 48003992 C T non_syn_coding MED 3 3 2_dad(dad;unaffected),2_mom(mom;affected),2_kid(child;affected) C/C,C/T,C/T 2_mom,2_kid 2
ASAH2C chr10 48004991 48004992 C T non_syn_coding MED 4 4 2_dad(dad;unaffected),2_mom(mom;affected),2_kid(child;affected) C/C,C/T,C/T 2_mom,2_kid 2
ASAH2C chr10 48003991 48003992 C T non_syn_coding MED 3 3 3_dad(dad;affected),3_mom(mom;unknown),3_kid(child;affected) C/T,C/C,C/T 3_dad,3_kid 2
ASAH2C chr10 48004991 48004992 C T non_syn_coding MED 4 4 3_dad(dad;affected),3_mom(mom;unknown),3_kid(child;affected) C/T,C/C,C/T 3_dad,3_kid 2
WDR37 chr10 1142207 1142208 T C stop_loss HIGH 1 1 2_dad(dad;unaffected),2_mom(mom;affected),2_kid(child;affected) T/T,T/C,T/C 2_mom,2_kid 2
WDR37 chr10 1142207 1142208 T C stop_loss HIGH 1 1 3_dad(dad;affected),3_mom(mom;unknown),3_kid(child;affected) T/C,T/T,T/C 3_dad,3_kid 2
x_linked_recessive: x-linked recessive inheritance¶
Note that as of version 0.19.0, we do not account for the pseudo autosomal regions. The ‘X’ chromosome can be specifing using –X (defaults to ‘chrX’ and ‘X’)
Genotype Requirements¶
- Affected females must be HOM_ALT
- Unaffected females are HET or HOM_REF
- Affected males are not HOM_REF
- Unaffected males are HOM_REF
x_linked_dominant: x-linked dominant inheritance¶
Note that as of version 0.19.0, we do not account for the pseudo autosomal regions. The ‘X’ chromosome can be specifing using –X (defaults to ‘chrX’ and ‘X’)
Genotype Requirements¶
- Affected males are HET or HOM_ALT
- Affected females must be HET
- Unaffecteds must be HOM_REF
- girls of affected dad must be affected
- boys of affected dad must be unaffected
- mothers of affected males must be het (and affected) [added in 0.19.1]
- at least 1 parent of affected females must be het (and affected). [added in 0.19.1]
x_linked_de_novo: x-linked de novo¶
Note that as of version 0.19.0, we do not account for the pseudo autosomal regions. The ‘X’ chromosome can be specifing using –X (defaults to ‘chrX’ and ‘X’)
Genotype Requirements¶
- affected female child must be het
- affected male child must be hom_alt (or het)
- parents should be unaffected and hom_ref
gene_wise: Custom genotype filtering by gene.¶
The gemini query tool allows querying by variant and the inheritance tools described above enable querying by gene for fixed inheritance patterns. The gene_wise tool allows querying by gene with custom genotype filters to bridge the gap between these tools.
With this tool, multiple –gt-filter s can be specified. Each filter can be any valid filter; often, it will make sense to have 1 filter for each family. For example, given this pedigree:
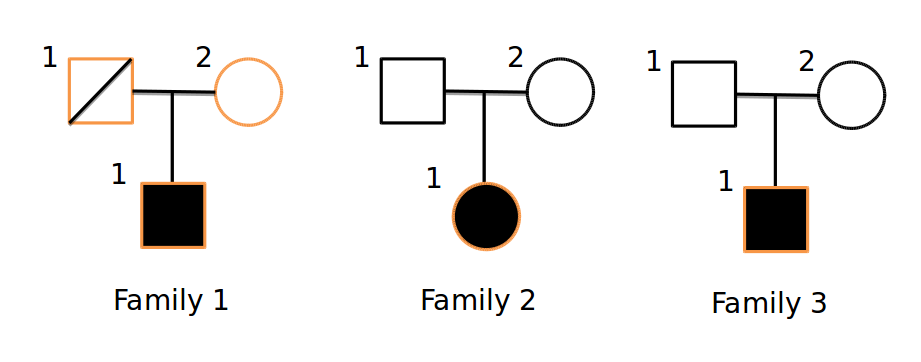
Where only the orange samples are sequenced, we could devise a query:
gemini gene_wise $db \
--min-filters 3 \
--gt-filter "gt_types.fam1_kid == HET and gt_types.fam1_mom == HOM_REF and gt_types.fam1_dad == HOM_REF" \
--gt-filter "gt_types.fam2_kid == HET" \
--gt-filter "gt_types.fam3_kid == HET" \
--columns "chrom,start,end,gene,impact,impact_severity" \
--filter "max_aaf_all < 0.005"
The –min-filters option means that we want all 3 of those filters to be met in a gene in order for variants in that gene to be reported. We can envision a scenario where we have 6 families (and 6 filters) and we want to report genes where 4 of them meet the filters. In that case, the query would have 6 –gt-filter s and –min-filters of 3.
This differs from using gemini query with a single –gt-filter that combines each of those terms with an and because this allows each filter to be met in a different variant but in the same gene while the gemini query tool applies all elements of the single filter to each variant.
The output from the above query is:
chrom start end gene impact impact_severity variant_filters n_gene_variants gene_filters
chr5 60839982 60839983 ZSWIM6 non_syn_coding MED 1,2,3 1 1,2,3
chr6 32548031 32548032 HLA-DRB1 non_syn_coding MED 1 4 1,2,3
chr6 32552059 32552060 HLA-DRB1 frame_shift HIGH 2 4 1,2,3
chr6 32552131 32552132 HLA-DRB1 inframe_codon_gain MED 3 4 1,2,3
chr6 32552136 32552137 HLA-DRB1 non_syn_coding MED 3 4 1,2,3
Note that the first gene has the same variant for all 3 families, so we could have found this with the gemini query tool. However, for the HLA gene, each of the 3 filters passed in different variant so this would be missed by the query tool which only looks at a single variant at a time.
As with the other tools, this tool orders by chromosome and gene and it applies WHERE (is_exonic = 1 AND impact_severity != ‘LOW’)” to the query.
- The variant_filters column shows which filters were passed by the variant.
- The n_gene_variants column shows how many variants in the gene are being reported.
- The gene_filter column shows which filters in the gene passed by any variant.
Multiple –gt-filter-required filters can also be specified. Each filter added to this argument is required to pass for each variant and it does not contribute to the –min-filters argument. This can be used with, or instead of –gt-filter E.g.
gemini gene_wise \
--columns "gene, chrom, start, end, ref, alt, impact, impact_severity" \
--gt-filter-required "((gt_depths).(*).(>10).(all))" \
--gt-filter "gt_types.fam1_kid == HET and gt_types.fam1_mom == HOM_REF and gt_types.fam1_dad == HOM_REF" \
--gt-filter "gt_types.fam2_kid == HET" \
--gt-filter "gt_types.fam3_kid == HET" \
--min-filters 2 \
test.db
will required that all samples meet the minimum depth filter and then keep the subset of those that meet 2 out of the 3 –gt-filters.
--where¶
By default gene_wise limits to variants passing the clause:
is_exonic = 1 AND impact_severity != ‘LOW’
but this can be changed with the –where clause, e.g.:
--where "(is_exonic = 1 or is_splicing = 1) AND impact_severity != 'LOW'"
pathways: Map genes and variants to KEGG pathways.¶
Mapping genes to biological pathways is useful in understanding the function/role played by a gene. Likewise, genes involved in common pathways is helpful in understanding heterogeneous diseases. We have integrated the KEGG pathway mapping for gene variants, to explain/annotate variation. This requires your VCF be annotated with either snpEff/VEP.
Examples:
$ gemini pathways -v 68 example.db
chrom start end ref alt impact sample genotype gene transcript pathway
chr10 52004314 52004315 T C intron M128215 C/C ASAH2 ENST00000395526 hsa00600:Sphingolipid_metabolism,hsa01100:Metabolic_pathways
chr10 126678091 126678092 G A stop_gain M128215 G/A CTBP2 ENST00000531469 hsa05220:Chronic_myeloid_leukemia,hsa04310:Wnt_signaling_pathway,hsa04330:Notch_signaling_pathway,hsa05200:Pathways_in_cancer
chr16 72057434 72057435 C T non_syn_coding M10475 C/T DHODH ENST00000219240 hsa01100:Metabolic_pathways,hsa00240:Pyrimidine_metabolism
Here, -v specifies the version of the Ensembl genes used to build the KEGG pathway map. Hence, use versions that match the VEP/snpEff versions of the annotated vcf for correctness. For e.g VEP v2.6 and snpEff v3.1 use Ensembl 68 version of the genomes.
We currently support versions 66 through 71 of the Ensembl genes
--lof¶
By default, all gene variants that map to pathways are reported. However,
one may want to restrict the analysis to LoF variants using the --lof option.
$ gemini pathways --lof -v 68 example.db
chrom start end ref alt impact sample genotype gene transcript pathway
chr10 126678091 126678092 G A stop_gain M128215 G/A CTBP2 ENST00000531469 hsa05220:Chronic_myeloid_leukemia,hsa04310:Wnt_signaling_pathway,hsa04330:Notch_signaling_pathway,hsa05200:Pathways_in_cancer
interactions: Find genes among variants that are interacting partners.¶
Integrating the knowledge of the known protein-protein interactions would be useful in explaining variation data. Meaning to say that a damaging variant in an interacting partner of a potential protein may be equally interesting as the protein itself. We have used the HPRD binary interaction data to build a p-p network graph which can be explored by GEMINI.
Examples:
$ gemini interactions -g CTBP2 -r 3 example.db
sample gene order_of_interaction interacting_gene
M128215 CTBP2 0_order: CTBP2
M128215 CTBP2 1_order: RAI2
M128215 CTBP2 2_order: RB1
M128215 CTBP2 3_order: TGM2,NOTCH2NL
Return CTBP2 (-g) interacting gene variants till the third order (-r)
lof_interactions¶
Use this option to restrict your analysis to only LoF variants.
$ gemini lof_interactions -r 3 example.db
sample lof_gene order_of_interaction interacting_gene
M128215 TGM2 1_order: RB1
M128215 TGM2 2_order: none
M128215 TGM2 3_order: NOTCH2NL,CTBP2
Meaning to say return all LoF gene TGM2 (in sample M128215) interacting partners to a 3rd order of interaction.
--var¶
An extended variant information (chrom, start, end etc.) for the interacting gene
may be achieved with the –var option for both the interactions and the
lof_interactions
$ gemini interactions -g CTBP2 -r 3 --var example.db
sample gene order_of_interaction interacting_gene var_id chrom start end impact biotype in_dbsnp clinvar_sig clinvar_disease_name aaf_1kg_all aaf_esp_all
M128215 CTBP2 0 CTBP2 5 chr10 126678091 126678092 stop_gain protein_coding 1 None None None None
M128215 CTBP2 1 RAI2 9 chrX 17819376 17819377 non_syn_coding protein_coding 1 None None 1 0.000473
M128215 CTBP2 2 RB1 7 chr13 48873834 48873835 upstream protein_coding 1 None None 0.94 None
M128215 CTBP2 3 NOTCH2NL 1 chr1 145273344 145273345 non_syn_coding protein_coding 1 None None None None
M128215 CTBP2 3 TGM2 8 chr20 36779423 36779424 stop_gain protein_coding 0 None None None None
$ gemini lof_interactions -r 3 --var example.db
sample lof_gene order_of_interaction interacting_gene var_id chrom start end impact biotype in_dbsnp clinvar_sig clinvar_disease_name aaf_1kg_all aaf_esp_all
M128215 TGM2 1 RB1 7 chr13 48873834 48873835 upstream protein_coding 1 None None 0.94 None
M128215 TGM2 3 NOTCH2NL 1 chr1 145273344 145273345 non_syn_coding protein_coding 1 None None None None
M128215 TGM2 3 CTBP2 5 chr10 126678091 126678092 stop_gain protein_coding 1 None None None None
lof_sieve: Filter LoF variants by transcript position and type¶
Not all candidate LoF variants are created equal. For e.g, a nonsense (stop gain) variant impacting the first 5% of a polypeptide is far more likely to be deleterious than one affecting the last 5%. Assuming you’ve annotated your VCF with snpEff v3.0+, the lof_sieve tool reports the fractional position (e.g. 0.05 for the first 5%) of the mutation in the amino acid sequence. In addition, it also reports the predicted function of the transcript so that one can segregate candidate LoF variants that affect protein_coding transcripts from processed RNA, etc.
$ gemini lof_sieve chr22.low.exome.snpeff.100samples.vcf.db
chrom start end ref alt highest_impact aa_change var_trans_pos trans_aa_length var_trans_pct sample genotype gene transcript trans_type
chr22 17072346 17072347 C T stop_gain W365* 365 557 0.655296229803 NA19327 C|T CCT8L2 ENST00000359963 protein_coding
chr22 17072346 17072347 C T stop_gain W365* 365 557 0.655296229803 NA19375 T|C CCT8L2 ENST00000359963 protein_coding
chr22 17129539 17129540 C T splice_donor None None None None NA18964 T|C TPTEP1 ENST00000383140 lincRNA
chr22 17129539 17129540 C T splice_donor None None None None NA19675 T|C TPTEP1 ENST00000383140 lincRNA
amend: updating / changing the sample information¶
Occassionally one may need to update the sample information
stored in the samples table. The amend tool allows one
to provide an updated PED file as input and it will update
each sample_id in the PED file that matches a sample_id.
For example, assume you have already loaded a GEMINI database
with a samples.ped where mom and dad are unaffected and
kid isaffected:
$ cat samples.ped
1 dad 0 0 1 1
1 mom 0 0 1 1
1 kid dad mom 2 2
$ gemini load -v my.vcf -p samples.ped -t VEP my.db
Now, let’s say you realized that the dad is also affected and you
want to correct the samples table accordingly. You would first
edit the PED file and then run the amend tool using the updated
PED.
$ cat samples.ped
1 dad 0 0 1 2
1 mom 0 0 1 1
1 kid dad mom 2 2
$ gemini amend --sample samples.ped my.db
annotate: adding your own custom annotations¶
It is inevitable that researchers will want to enhance the gemini framework with
their own, custom annotations. gemini provides a sub-command called
annotate for exactly this purpose. As long as you provide a tabix‘ed
annotation file in BED or VCF format, the annotate tool will, for each
variant in the variants table, screen for overlaps in your annotation file and
update a one or more new column in the variants table that you may specify on the command
line. This is best illustrated by example.
Let’s assume you have already created a gemini database of a VCF file using
the load module.
$ gemini load -v my.vcf -t snpEff my.db
Now, let’s imagine you have an annotated file in BED format (important.bed)
that describes regions of the genome that are particularly relevant to your
lab’s research. You would like to annotate in the gemini database which variants
overlap these crucial regions. We want to store this knowledge in a new column
in the variants table called important_variant that tracks whether a given
variant overlapped (1) or did not overlap (0) intervals in your annotation file.
To do this, you must first TABIX your BED file:
$ bgzip important.bed
$ tabix -p bed important.bed.gz
-a boolean Did a variant overlap a region or not?¶
Note
Formerly, the -a option was the -t option.
Now, you can use this TABIX’ed file to annotate which variants overlap your
important regions. In the example below, the results will be stored in a new
column called “important”. The -t boolean option says that you just want to
track whether (1) or not (0) the variant overlapped one or more of your regions.
$ gemini annotate -f important.bed.gz -c important -a boolean my.db
Since a new columns has been created in the database, we can now directly query the new column. In the example results below, the first and third variants overlapped a crucial region while the second did not.
$ gemini query \
-q "select chrom, start, end, variant_id, important from variants" \
my.db \
| head -3
chr22 100 101 1 1
chr22 200 201 2 0
chr22 300 500 3 1
-a count How many regions did a variant overlap?¶
Instead of a simple yes or no, we can use the -t count option to count
how many important regions a variant overlapped. It turns out that the 3rd
variant actually overlapped two important regions.
$ gemini annotate -f important.bed.gz -c important -a count my.db
$ gemini query \
-q "select chrom, start, end, variant_id, crucial from variants" \
my.db \
| head -3
chr22 100 101 1 1
chr22 200 201 2 0
chr22 300 500 3 2
-a extract Extract specific values from a BED file¶
Lastly, we may also extract values from specific fields in a BED
file (or from the INFO field in a VCF) and populate one or more new columns
in the database based on
overlaps with the annotation file and the values of the fields therein.
To do this, we use the -a extract option.
This is best described with an example. To set this up, let’s imagine that we have a VCF file from a different experiment and we want to annotate the variants in our GEMINI database with the allele frequency and depth tags from the INFO fields for the same variants in this other VCF file.
# bgzip and tabix the vcf for use with the annotate tool. $ bgzip other.vcf $ tabix other.vcf.gz
Now that we have a proper TABIX’ed VCF file, we can use the -a extract option to populate new
columns in the GEMINI database. In order to do so, we must specify:
- its type (e.g., text, int, float,) (
-t)- the field in the INFO column of the VCF file that we should use to extract data with which to populate the new column (
-e)- what operation should be used to summarize the data in the event of multiple overlaps in the annotation file (
-o)- (optionally) the name of the column we want to add (
-c), if this is not specified, it will use the value from-e.
For example, let’s imagine we want to create a new column called “other_allele_freq” using the AF field in our VCF file to populate it.
$ gemini annotate -f other.vcf.gz \
-a extract \
-c other_allele_freq \
-t float \
-e AF \
-o mean \
my.db
This create a new column in my.db called other_allele_freq and this
new column will be a FLOAT. In the event of multiple records in the VCF
file overlapping a variant in the database, the average (mean) of the allele
frequencies values from the VCF file will be used.
At this point, one can query the database based on the values of the
new other_allele_freq column:
$ gemini query -q "select * from variants where other_allele_freq < 0.01" my.db
-t TYPE Specifying the column type(s) when using -a extract¶
The annotate tool will create three different types of columns via the -t option:
- Floating point columns for annotations with decimal precision as above (
-t float)- Integer columns for integral annotations (
-t integer)- Text columns for string columns such as “valid”, “yes”, etc. (
-t text)
Note
The -t option is only valid when using the -a extract option.
-o OPERATION Specifying the summary operations when using -a extract¶
In the event of multiple overlaps between a variant and records in the annotation
file, the annotate tool can summarize the values observed with multiple options:
-o mean. Compute the average of the values. They must be numeric.-o median. Compute the median of the values. They must be numeric.-o min. Compute the minimum of the values. They must be numeric.-o max. Compute the maximum of the values. They must be numeric.-o mode. Compute the maximum of the values. They must be numeric.-o first. Use the value from the first record in the annotation file.-o last. Use the value from the last record in the annotation file.-o list. Create a comma-separated list of the observed values. -t must be text-o uniq_list. Create a comma-separated list of the distinct (i.e., non-redundant) observed values. -t must be text-o sum. Compute the sum of the values. They must be numeric.
Note
The -o option is only valid when using the -a extract option.
Annotating with VCF¶
Most of the examples to this point have pulled a column from a tabix indexed bed file.
It is likewise possible to pull from the INFO field of a tabix index VCF. The syntax
is identical but the -e operation will specify the names of fields in the INFO column
to pull. By default, those names will be used, but that can still be specified with the
-c column.
Here are some example uses
# put a DP column in the db:
gemini annotate -f anno.vcf.gz -o list -e DP -t integer my.db
# ... and name it 'depth'
gemini annotate -f anno.vcf.gz -o list -e DP -c depth -t integer my.db
# use multiple columns
gemini annotate -f anno.vcf.gz -o list,mean -e DP,Qmeter -c depth,qmeter -t integer my.db
Missing values are allowed since we expect that in some cases an annotation VCF will not have all INFO fields specified for all variants.
Note
We recommend decomposing and normalizing variants before annotating. See Step 1. split, left-align, and trim variants for a detailed explanation of how to do this.
Extracting and populating multiple columns at once.¶
One can also extract and populate multiple columns at once by providing
comma-separated lists (no spaces) of column names (-c), types (-t), numbers (-e),
and summary operations (-o). For example, recall that in the VCF example above,
we created a TABIX’ed BED file containg the allele frequency and depth values from
the INFO field as the 4th and 5th columns in the BED, respectively.
Instead of running the annotate tool twice (once for eaxh column), we can
run the tool once and load both columns in the same run. For example:
$ gemini annotate -f other.bed.gz \
-a extract \
-c other_allele_freq,other_depth \
-t float,integer \
-e 4,5 \
-o mean,max \
my.db
We can then use each of the new columns to filter variants with a GEMINI query:
$ gemini query -q "select * from variants \
where other_allele_freq < 0.01 \
and other_depth > 100" my.db
region: Extracting variants from specific regions or genes¶
One often is concerned with variants found solely in a particular gene or
genomic region. gemini allows one to extract variants that fall within
specific genomic coordinates as follows:
--reg¶
$ gemini region --reg chr1:100-200 my.db
--gene¶
Or, one can extract variants based on a specific gene name.
$ gemini region --gene PTPN22 my.db
--columns¶
By default, this tool reports all columns in the variants table. One may
choose to report only a subset of the columns using the --columns option. For
example, to report just the gene, chrom, start, end, ref, alt, impact, and impact_severity columns, one
would use the following:
$ gemini region --gene DHODH \
--columns "chrom, start, end, ref, alt, gene, impact" \
my.db
chr16 72057281 72057282 A G DHODH intron
chr16 72057434 72057435 C T DHODH non_syn_coding
chr16 72059268 72059269 T C DHODH downstream
--filter¶
By default, this tool will report all variants regardless of their putative
functional impact. In order to apply additional constraints on the variants
returned, one can use the --filter option. Using SQL syntax, conditions
applied with the ``–filter option become WHERE clauses in the query issued to
the GEMINI database. For example, if we wanted to restrict candidate variants
to solely those with a HIGH predicted functional consequence, we could use the
following:
$ gemini region --gene DHODH \
--columns "chrom, start, end, ref, alt, gene, impact" \
--filter "alt='G'"
my.db
chr16 72057281 72057282 A G DHODH intron
--json¶
Reporting query output in JSON format may enable HTML/Javascript apps to query GEMINI and retrieve the output in a format that is amenable to web development protocols.
To report in JSON format, use the --json option. For example:
$ gemini region --gene DHODH \
--columns "chrom, start, end, ref, alt, gene, impact" \
--filter "alt='G'"
--json
my.db
{"chrom": "chr16", "start": 72057281, "end": 72057282, "ref": "A", "alt": "G", "gene": "DHODH"}
windower: Conducting analyses on genome “windows”.¶
gemini includes a convenient tool for computing variation metrics across
genomic windows (both fixed and sliding). Here are a few examples to whet your
appetite. If you’re still hungry, contact us.
Compute the average nucleotide diversity for all variants found in non-overlapping, 50Kb windows.
$ gemini windower -w 50000 -s 0 -t nucl_div -o mean my.db
Compute the average nucleotide diversity for all variants found in 50Kb windows that overlap by 10kb.
$ gemini windower -w 50000 -s 10000 -t nucl_div -o mean my.db
Compute the max value for HWE statistic for all variants in a window of size 10kb
$ gemini windower -w 10000 -t hwe -o max my.db
stats: Compute useful variant statistics.¶
The stats tool computes some useful variant statistics like
Compute the transition and transversion ratios for the snps
$ gemini stats --tstv my.db
ts tv ts/tv
4 5 0.8
--tstv-coding¶
Compute the transition/transversion ratios for the snps in the coding regions.
--tstv-noncoding¶
Compute the transition/transversion ratios for the snps in the non-coding regions.
Compute the type and count of the snps.
$ gemini stats --snp-counts my.db
type count
A->G 2
C->T 1
G->A 1
Calculate the site frequency spectrum of the variants.
$ gemini stats --sfs my.db
aaf count
0.125 2
0.375 1
Compute the pair-wise genetic distance between each sample
$ gemini stats --mds my.db
sample1 sample2 distance
M10500 M10500 0.0
M10475 M10478 1.25
M10500 M10475 2.0
M10500 M10478 0.5714
Return a count of the types of genotypes per sample
$ gemini stats --gts-by-sample my.db
sample num_hom_ref num_het num_hom_alt num_unknown total
M10475 4 1 3 1 9
M10478 2 2 4 1 9
Return the total variants per sample (sum of homozygous and heterozygous variants)
$ gemini stats --vars-by-sample my.db
sample total
M10475 4
M10478 6
--summarize¶
If none of these tools are exactly what you want, you can summarize the variants per sample of an arbitrary query using the –summarize flag. For example, if you wanted to know, for each sample, how many variants are on chromosome 1 that are also in dbSNP:
$ gemini stats --summarize "select * from variants where in_dbsnp=1 and chrom='chr1'" my.db
sample total num_het num_hom_alt
M10475 1 1 0
M128215 1 1 0
M10478 2 2 0
M10500 2 1 1
burden: perform sample-wise gene-level burden calculations¶
The burden tool provides a set of utilities to perform burden
summaries on a per-gene, per sample basis. By default, it outputs
a table of gene-wise counts of all high impact variants in coding regions for
each sample:
$ gemini burden test.burden.db
gene M10475 M10478 M10500 M128215
WDR37 2 2 2 2
CTBP2 0 0 0 1
DHODH 1 0 0 0
--nonsynonymous¶
If you want to be a little bit less restrictive, you can include all non-synonymous variants instead:
$ gemini burden --nonsynonymous test.burden.db
gene M10475 M10478 M10500 M128215
SYCE1 0 1 1 0
WDR37 2 2 2 2
CTBP2 0 0 0 1
ASAH2C 2 1 1 0
DHODH 1 0 0 0
--calpha¶
If your database has been loaded with a PED file describing case and control samples, you can calculate the c-alpha statistic for cases vs. control:
$ gemini burden --calpha test.burden.db
gene T c Z p_value
SYCE1 -0.5 0.25 -1.0 0.841344746069
WDR37 -1.0 1.5 -0.816496580928 0.792891910879
CTBP2 0.0 0.0 nan nan
ASAH2C -0.5 0.75 -0.57735026919 0.718148569175
DHODH 0.0 0.0 nan nan
To calculate the P-value using a permutation test, use the --permutations option,
specifying the number of permutations of the case/control labels you want to use.
--min-aaf and --max-aaf for --calpha¶
By default, all variants affecting a given gene will be included in the
C-alpha computation. However, one may establish alternate allele frequency
boundaries for the variants included using the --min-aaf and
--max-aaf options.
$ gemini burden --calpha test.burden.db --min-aaf 0.0 --max-aaf 0.01
--cases and --controls for ``--calpha¶
If you do not have a PED file loaded, or your PED file does not follow the standard PED phenotype encoding format you can still perform the c-alpha test, but you have to specify which samples are the control samples and which are the case samples:
$ gemini burden --controls M10475 M10478 --cases M10500 M128215 --calpha test.burden.db
gene T c Z p_value
SYCE1 -0.5 0.25 -1.0 0.841344746069
WDR37 -1.0 1.5 -0.816496580928 0.792891910879
CTBP2 0.0 0.0 nan nan
ASAH2C -0.5 0.75 -0.57735026919 0.718148569175
DHODH 0.0 0.0 nan nan
--nonsynonymous --calpha¶
If you would rather consider all nonsynonymous variants for the C-alpha test rather
than just the medium and high impact variants, add the --nonsynonymous flag.
ROH: Identifying runs of homozygosity¶
Runs of homozygosity are long stretches of homozygous genotypes that reflect segments shared identically by descent and are a result of consanguinity or natural selection. Consanguinity elevates the occurrence of rare recessive diseases (e.g. cystic fibrosis) that represent homozygotes for strongly deleterious mutations. Hence, the identification of these runs holds medical value.
The ‘roh’ tool in GEMINI returns runs of homozygosity identified in whole genome data. The tool basically looks at every homozygous position on the chromosome as a possible start site for the run and looks for those that could give rise to a potentially long stretch of homozygous genotypes.
For e.g. for the given example allowing 1 HET genotype (h) and 2 UKW genotypes (u)
the possible roh runs (H) would be:
genotype_run = H H H H h H H H H u H H H H H u H H H H H H H h H H H H H h H H H H H
roh_run1 = H H H H h H H H H u H H H H H u H H H H H H H
roh_run2 = H H H H u H H H H H u H H H H H H H h H H H H H
roh_run3 = H H H H H u H H H H H H H h H H H H H
roh_run4 = H H H H H H H h H H H H H
roh returned for –min-snps = 20 would be:
roh_run1 = H H H H h H H H H u H H H H H u H H H H H H H
roh_run2 = H H H H u H H H H H u H H H H H H H h H H H H H
As you can see, the immediate homozygous position right of a break (h or u) would be the possible start of a new roh run and genotypes to the left of a break are pruned since they cannot be part of a longer run than we have seen before.
Return roh with minimum of 50 snps, a minimum run length of 1 mb and a minimum sample depth of 20
for sample S138 (with default values for allowed number of HETS, UNKS and total depth).
$ gemini roh --min-snps 50 \
--min-gt-depth 20 \
--min-size 1000000 \
-s S138 \
roh_run.db
chrom start end sample num_of_snps density_per_kb run_length_in_bp
chr2 233336080 234631638 S138 2583 1.9953 1295558
chr2 238341281 239522281 S138 2899 2.4555 1181000
set_somatic: Flag somatic variants¶
Somatic mutations in a tumor-normal pair are variants that are present in the tumor but not in the normal sample.
Note
1. This tool requires that you specify the sample layout via a PED file when loading your VCF into GEMINI via:
gemini load -v my.vcf -p my.ped my.db
Example PED file format for GEMINI
#Family_ID Individual_ID Paternal_ID Maternal_ID Sex Phenotype Ethnicity
1 Normal -9 -9 0 1 -9
1 Tumor -9 -9 0 2 -9
default behavior¶
By default, set_somatic simply marks variants that are genotyped as
homozygous reference in the normal sample and non-reference in the tumor.
More stringent somatic filtering criteria are available through tunable
command line parameters.
$ gemini set_somatic \
--min-depth 30 \
--min-qual 20 \
--min-somatic-score 18 \
--min-tumor-depth 10 \
--min-norm-depth 10 \
tumor_normal.db
tum_name tum_gt tum_alt_freq tum_alt_depth tum_depth nrm_name nrm_gt nrm_alt_freq nrm_alt_depth nrm_depth chrom start end ref alt gene
tumor GAAAAAAAAAAAAAGGTGAAAATT/GAAAAAAAAAAAAGGTGAAAATT 0.217391304348 5 23 normal GAAAAAAAAAAAAAGGTGAAAATT/GAAAAAAAAAAAAAGGTGAAAATT 0.0 0 25 chrX 132838304 132838328 GAAAAAAAAAAAAAGGTGAAAATT GAAAAAAAAAAAAGGTGAAAATT GPC3
tumor CTGCTATTTTG/CG 0.22 11 50 normal CTGCTATTTTG/CTGCTATTTTG 0.0 0 70 chr17 59861630 59861641 CTGCTATTTTG CG BRIP1
tumor C/A 0.555555555556 10 18 normal C/C 0.0 0 17 chr17 7578460 7578461 C A TP53
tumor C/T 0.1875 12 64 normal C/C 0.0 0 30 chr2 128046288 128046289 C T ERCC3
Identified and set 4 somatic mutations
--min-depth [None]¶
The minimum required combined depth for tumor and normal samples.
--min-qual [None]¶
The minimum required variant quality score.
--min-somatic-score [None]¶
The minimum required somatic score (SSC). This score is produced by various somatic variant detection algorithms including SpeedSeq, SomaticSniper, and VarScan 2.
--max-norm-alt-freq [None]¶
The maximum frequency of the alternate allele allowed in the normal sample.
--max-norm-alt-count [None]¶
The maximum count of the alternate allele allowed in the normal sample.
--min-norm-depth [None]¶
The minimum depth required in the normal sample.
--min-tumor-alt-freq [None]¶
The minimum frequency of the alternate allele required in the tumor sample.
--min-tumor-alt-count [None]¶
The minimum count of the alternate allele required in the tumor sample.
--min-tumor-depth [None]¶
The minimum depth required in the tumor sample.
--chrom [None]¶
A specific chromosome on which to flag somatic mutations.
--dry-run¶
Don’t set the is_somatic flag, just report what _would_ be set. For testing purposes.
actionable_mutations: Report actionable somatic mutations and drug-gene interactions¶
Actionable mutations are somatic variants in COSMIC cancer census genes with medium or high impact severity predictions. This tool reports actionable mutations as well as their known drug interactions (if any) from DGIdb. Current functionality is only for SNVs and indels.
Note
- This tool requires somatic variants to have been flagged using
set_somatic
$ gemini actionable_mutations tumor_normal.db
tum_name chrom start end ref alt gene impact is_somatic in_cosmic_census dgidb_info
tumor chr2 128046288 128046289 C T ERCC3 non_syn_coding 1 1 None
tumor chr17 7578460 7578461 C A TP53 non_syn_coding 1 1 {'searchTerm': 'TP53', 'geneCategories': ['CLINICALLY ACTIONABLE', 'DRUGGABLE GENOME', 'TUMOR SUPPRESSOR', 'TRANSCRIPTION FACTOR COMPLEX', 'DRUG RESISTANCE', 'HISTONE MODIFICATION', 'DNA REPAIR', 'TRANSCRIPTION FACTOR BINDING'], 'geneName': 'TP53', 'geneLongName': 'tumor protein p53', 'interactions': [{'source': 'DrugBank', 'interactionId': '711cbe42-4930-4b46-963e-79ab35bbbd0f', 'interactionType': 'n/a', 'drugName': '1-(9-ETHYL-9H-CARBAZOL-3-YL)-N-METHYLMETHANAMINE'}, {'source': 'PharmGKB', 'interactionId': '8234d9b9-085d-49b1-aac2-cf5375d91477', 'interactionType': 'n/a', 'drugName': 'FLUOROURACIL'}, {'source': 'PharmGKB', 'interactionId': '605d7bca-7ed9-428e-aa7c-f76aafd66b54', 'interactionType': 'n/a', 'drugName': 'PACLITAXEL'}, {'source': 'TTD', 'interactionId': '1fe9db63-3581-435b-b22a-12d45c8c9864', 'interactionType': 'activator', 'drugName': 'CURAXIN CBLC102'}, {'source': 'TALC', 'interactionId': '8f8f6822-cb9e-40aa-8360-5532e059f1e7', 'interactionType': 'vaccine', 'drugName': 'EP-2101'}, {'source': 'TALC', 'interactionId': 'd59e14bc-b9a5-4c9f-a5aa-7ba322f0fa0e', 'interactionType': 'vaccine', 'drugName': 'MUTANT P53 PEPTIDE PULSED DENDRITIC CELL'}, {'source': 'TALC', 'interactionId': '79256b6e-9a16-4fbe-a237-28dbca28bc2a', 'interactionType': 'vaccine', 'drugName': 'AD.P53-DC'}]}
tumor chr17 59861630 59861641 CTGCTATTTTG CG BRIP1 inframe_codon_loss 1 1 None
tumor chrX 132838304 132838328 GAAAAAAAAAAAAAGGTGAAAATT GAAAAAAAAAAAAGGTGAAAATT GPC3 splice_region 1 1 None
fusions: Report putative gene fusions¶
Report putative somatic gene fusions from structural variants in a tumor-normal pair. Putative fusions join two genes and preserve transcript strand orientation.
Note
- This tool requires somatic variants to have been flagged using
set_somatic
default behavior¶
By default, fusions reports structural variants that are flagged as
somatic, join two different genes, and preserve transcript strand orientation.
These may be further filtered using tunable command line parameters.
$ gemini fusions \
--min_qual 5 \
--in_cosmic_census \
tumor_normal.db
chromA breakpointA_start breakpointA_end chromB breakpointB_start breakpointB_end var_id qual strandA strandB sv_type geneA geneB tool evidence_type is_precise sample
chr3 176909953 176909982 chr3 178906001 178906030 1233 9.58 - + complex TBL1XR1 PIK3CA LUMPY PE 0 tumor
--min_qual [None]¶
The minimum required variant quality score.
--evidence_type STRING¶
The required supporting evidence types for the variant from LUMPY (“PE”, “SR”, or “PE,SR”).
--in_cosmic_census¶
Require at least one of the affected genes to be in the COSMIC cancer gene census.
db_info: List the gemini database tables and columns¶
Because of the sheer number of annotations that are stored in gemini, there are
admittedly too many columns to remember by rote. If you can’t recall the name of
particular column, just use the db_info tool. It will report all of the
tables and all of the columns / types in each table:
$ gemini db_info test.db
table_name column_name type
variants chrom text
variants start integer
variants end integer
variants variant_id integer
variants anno_id integer
variants ref text
variants alt text
variants qual float
variants filter text
variants type text
variants sub_type text
variants gts blob
variants gt_types blob
variants gt_phases blob
variants gt_depths blob
variants call_rate float
variants in_dbsnp bool
variants rs_ids text
variants in_omim bool
variants clin_sigs text
variants cyto_band text
variants rmsk text
variants in_cpg_island bool
variants in_segdup bool
variants is_conserved bool
variants num_hom_ref integer
variants num_het integer
variants num_hom_alt integer
variants num_unknown integer
variants aaf float
variants hwe float
variants inbreeding_coeff float
variants pi float
variants recomb_rate float
variants gene text
variants transcript text
variants is_exonic bool
variants is_coding bool
variants is_lof bool
variants exon text
variants codon_change text
variants aa_change text
variants aa_length text
variants biotype text
variants impact text
variants impact_severity text
variants polyphen_pred text
variants polyphen_score float
variants sift_pred text
variants sift_score float
variants anc_allele text
variants rms_bq float
variants cigar text
variants depth integer
variants strand_bias float
variants rms_map_qual float
variants in_hom_run integer
variants num_mapq_zero integer
variants num_alleles integer
variants num_reads_w_dels float
variants haplotype_score float
variants qual_depth float
variants allele_count integer
variants allele_bal float
variants in_hm2 bool
variants in_hm3 bool
variants is_somatic
variants in_esp bool
variants aaf_esp_ea float
variants aaf_esp_aa float
variants aaf_esp_all float
variants exome_chip bool
variants in_1kg bool
variants aaf_1kg_amr float
variants aaf_1kg_asn float
variants aaf_1kg_afr float
variants aaf_1kg_eur float
variants aaf_1kg_all float
variants grc text
variants gms_illumina float
variants gms_solid float
variants gms_iontorrent float
variants encode_tfbs
variants encode_consensus_gm12878 text
variants encode_consensus_h1hesc text
variants encode_consensus_helas3 text
variants encode_consensus_hepg2 text
variants encode_consensus_huvec text
variants encode_consensus_k562 text
variants encode_segway_gm12878 text
variants encode_segway_h1hesc text
variants encode_segway_helas3 text
variants encode_segway_hepg2 text
variants encode_segway_huvec text
variants encode_segway_k562 text
variants encode_chromhmm_gm12878 text
variants encode_chromhmm_h1hesc text
variants encode_chromhmm_helas3 text
variants encode_chromhmm_hepg2 text
variants encode_chromhmm_huvec text
variants encode_chromhmm_k562 text
variant_impacts variant_id integer
variant_impacts anno_id integer
variant_impacts gene text
variant_impacts transcript text
variant_impacts is_exonic bool
variant_impacts is_coding bool
variant_impacts is_lof bool
variant_impacts exon text
variant_impacts codon_change text
variant_impacts aa_change text
variant_impacts aa_length text
variant_impacts biotype text
variant_impacts impact text
variant_impacts impact_severity text
variant_impacts polyphen_pred text
variant_impacts polyphen_score float
variant_impacts sift_pred text
variant_impacts sift_score float
samples sample_id integer
samples name text
samples family_id integer
samples paternal_id integer
samples maternal_id integer
samples sex text
samples phenotype text
samples ethnicity text
